Translational Research
Accelerating the clinical application of promising research findings

Home » Translational Research » Research Highlights » Here
In this section
American Society of Human Genetics 2010 annual conference
Following are highlights of five Scripps Translational Science Institute (STSI) presentations at the conference, which was held Nov. 2 to 6, in Washington, D.C.
In a platform presentation titled, “Genome-wide association of bipolar disorder suggests an enrichment of true associations in regions near exons,” STSI Research Associate Erin Smith, Ph.D., summarized the results of an analysis of genome-wide association study (GWAS) datasets on 2,191 patients with bipolar disorder (BD) and 1,434 controls. European ancestry characterized both patients and controls in the datasets, which were provided by the Genetic Association Information Network (GAIN) and the Translational Genomics Research Institute (TGen).
She and her colleagues did not identify an association that individually reached genome-wide significance in the GAIN and TGen studies or in a meta-analysis of the Wellcome Trust Case Control Consortium (WTCCC) BD data. However, by focusing on the single nucleotide polymorphisms (SNPs) that the WTCCC BD study had identified as having the strongest associations, they found an enrichment of associations at p<0.05 in the SNPs with the highest power.
Shared underlying genetics for BD exist between the WTCCC and the GAIN-TGen studies, according to the analysis by Dr. Smith and her collaborators. They also determined that the SNPS that are more likely to replicate are:
- high powered—indicating that more samples are likely to have significant regions; and
- located near genes and showing the same direction of effect.
Despite a lack of genome-wide significance for any individual variant in a single large study, analysis of groups of SNPs that showed consistent effects across studies can provide insight about the underlying genetic mechanism of BD, said Dr. Smith, whose collaborators included: Nicholas J. Schork, Ph.D., of STSI; David Craig, Ph.D., of TGen; John Kelsoe, M.D., of UC at San Diego and the V.A. San Diego Healthcare System; and the Bipolar Genome Study Consortium, which also includes STSI scientists Cinnamon S. Bloss, Ph.D., and Sarah S. Murray, Ph.D.
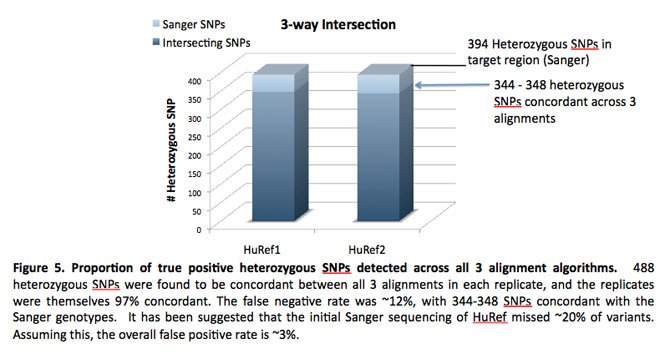
Ashley Van Zeeland, Ph.D., presented a poster entitled, “Comparison of Sequence Alignment Algorithms for Targeted Sequencing Studies,” about using real data from a targeted sequencing study to compare the performance of both hash-based and Burrows-Wheeler transform (BWT) alignment methods. The STSI study likely is the first algorithm comparison that did not use simulated data.
A Dickinson Research Fellow at STSI, Dr. Van Zeeland said that the results are relevant to translational genomics because the accurate alignment of sequencing “reads” to the reference genome is one of the fundamental steps in the analysis of human sequence data. All subsequent analyses, from calling single nucleotide polymorphisms (SNPs) and insertion/deletions (indels) to statistical analyses of variants, rely on accurate and high-quality alignment of the sequencing reads. Moreover, alignment results can provide the first measure of the success of the sequencing experiment by determining the effectiveness of the library preparation, and if applicable, the target enrichment.
Dr. Van Zeeland and her collaborators targeted about 1.3Mb of coding sequence in 150 candidate genes. To accurately assess the relative success of each of the alignment algorithms, they mapped the reads from the “gold standard,” fully sequenced HuRef DNA sample back to the reference genome. Preliminary results indicated that all mapping algorithms attained an acceptable level of mapped reads and achieved high levels of concordance with the HuRef genome. However, the authors noted that using multiple alignment methods and then only using those variants called by more than one method may result in the most accurate variant calls from sequence data.
Dr. Van Zeeland’s collaborators included Tierney Phillips, Marian Shaw, Nicholas J. Schork, Ph.D., and Samuel Levy, Ph.D., of STSI; and Paul Hoover, Ph.D., Wayne Pfeiffer, Ph.D., and Mark Miller, Ph.D., of the San Diego Supercomputer Center.
Sarah S. Murray, Ph.D., Director of Genetics at STSI, presented a poster titled, “Sequence Variants in Translation Regulator EIF4EBP3 Gene Region Associate with Disease Susceptibility,” about ongoing research to understand the genes and pathways related to health span.
Dr. Murray and her colleagues genotyped over one million SNPs in the DNA samples of 397 participants in the “Wellderly” study and 386 individuals (“illderly” control group) who had died from various chronic illnesses and were matched for birth year, gender and ancestry with the “Wellderly” participants. In a GWAS of the whole-genome genotyping data on these two groups, the researchers found that multiple SNPs in the translator regulator EIF4EBP3 were associated where the allele frequency was higher in the “illderly” control group versus the “Wellderly” participants. EIF4EBP3 with EIF4EBP1 and EIF4EBP2 produce proteins that bind to eIF4E. The binding of these proteins to eIF4G inhibits translation and affects cell proliferation.
Dr. Murray’s collaborators include Bradley Patay, M.D., of Scripps Health and STSI; Erin Smith, Ph.D., Cinnamon Bloss, Ph.D., Ali Torkamani, Ph.D., Nikki Villarasa, Nicholas J. Schork, Ph.D., Samuel Levy, Ph.D., and Eric J. Topol, M.D., of STSI; Kai Wang, Ph.D., of Children’s Hospital of Philadelphia; Eric Orwoll, M.D., of Oregon Health Sciences University; and Steven Cummings, M.D., of California Pacific Medical Center.
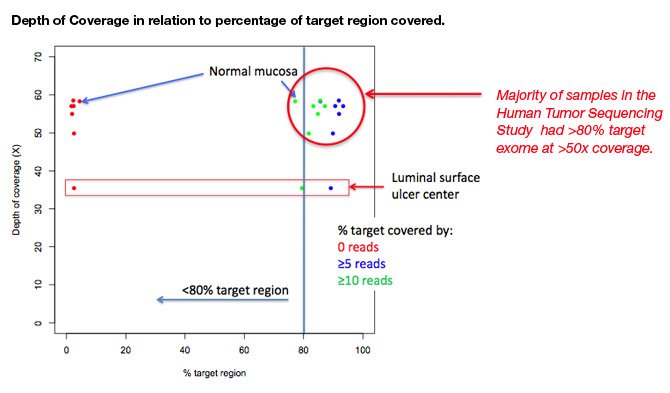
Samuel Levy, Ph.D., Director of Genome Sciences at STSI, presented a poster titled, “The role of genomic heterogeneity in primary tumors from colon cancer patients,” about findings from STSI’s ongoing Human Tumor Sequencing study.
In different anatomic areas of one patient’s colon cancer, Dr Levy and his collaborators at The Scripps Research Institute (TSRI) and Scripps Clinic used a genomics based approach to characterize gene-based mutational events common and unique to each anatomic locus in a tumor. To understand how a precise molecular characterization of events within a single tumor is influenced by the tumor’s heterogeneity, Dr. Levy and his colleagues developed a methodology that measured and established the relationship between changes in the exome in relation to tumor locus (epicenter, invading edge, and tumor to normal tissue transition).
Dr. Levy’s collaborators include Glenn Oliveira, Andrew Carson, Ph.D., and Ali Torkamani, Ph.D., of STSI; Kelly Bethel, M.D., of Scripps Clinic; and Peter Kuhn, Ph.D., of TSRI.
Nicholas J. Schork, Ph.D., was the senior author of the platform presentation titled, “Power of equal intercepts differential effect regression models to infer gene-environment interactions in genome-wide association studies.”
The presentation, given by Dr. Schork’s collaborator, Guia Guffanti, Ph.D., of the University of California at Irvine (UCI), reported research relevant to understanding the genetic variants that contribute differentially to quantitative phenotypes in the wide ranging genetic or environmental backgrounds that mediate the biology of complex diseases.
Dr. Schork and his collaborators studied a simple differential effects regression model that assumed equal intercepts for detecting specific types of gene x environment interactions on quantitative traits in the context of genome-wide association studies. The presentation described the results of simulation studies for different settings that resulted in both the characterization of gene x environment interactions as well as an increased power to detect them in comparison with the traditional linear model.
Intuitive and easy to implement statistical analysis models are important since gene x environment interactions are known to influence common complex diseases but are not often easy to detect because of the myriad ways in which both environmental factors and genes may interact.
In addition to Drs. Schork and Guffanti, co-authors included: Trygve Bakken of STSI; Steven Potkin, Ph.D., of UCI; and Fabio Macciardi, Ph.D., of the University of Milan in Italy.